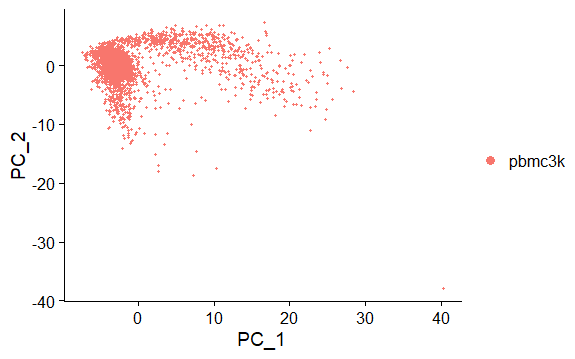
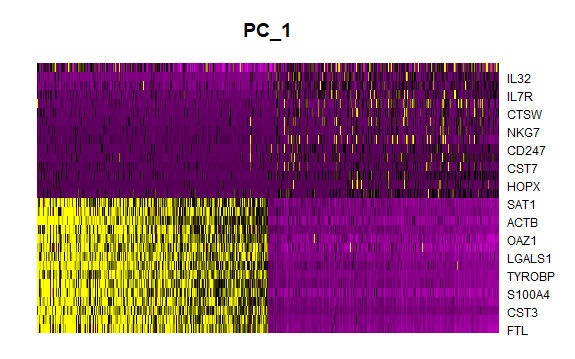
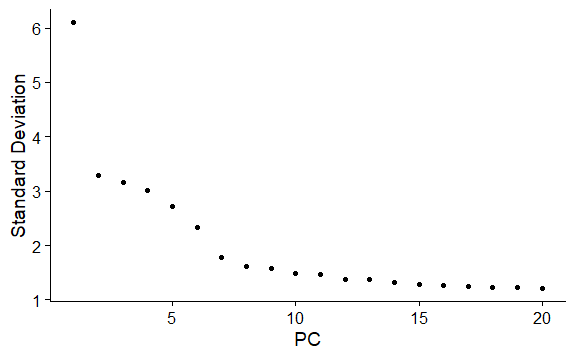
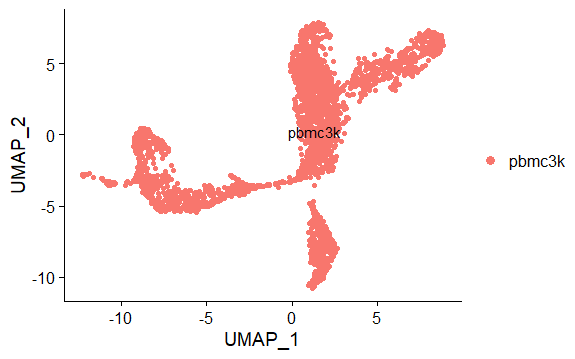
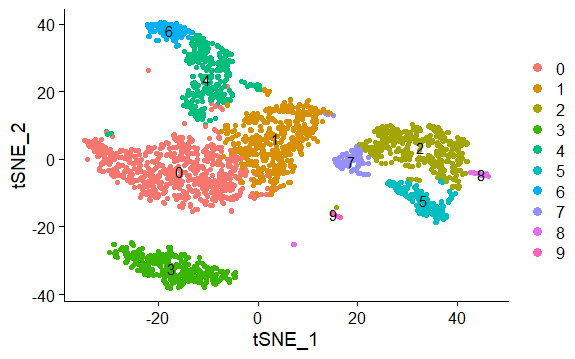
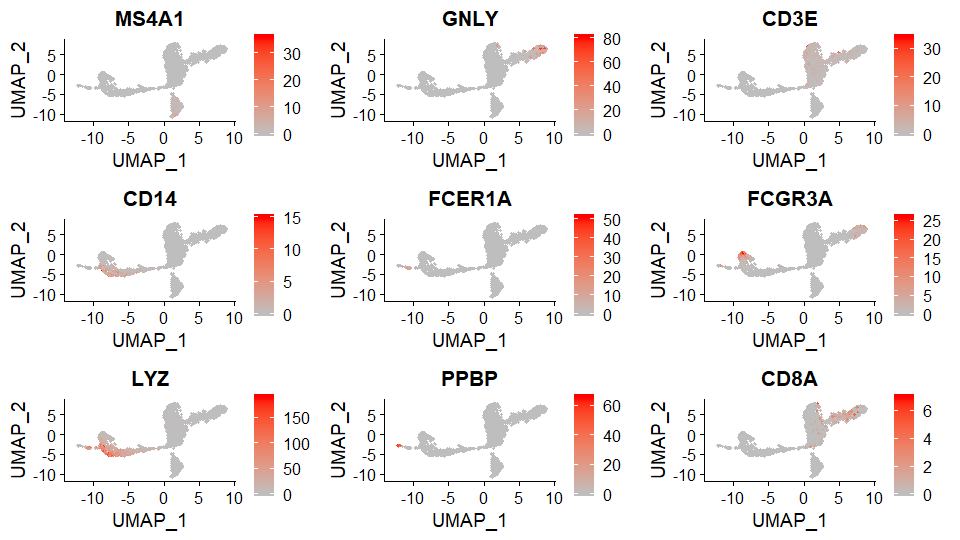
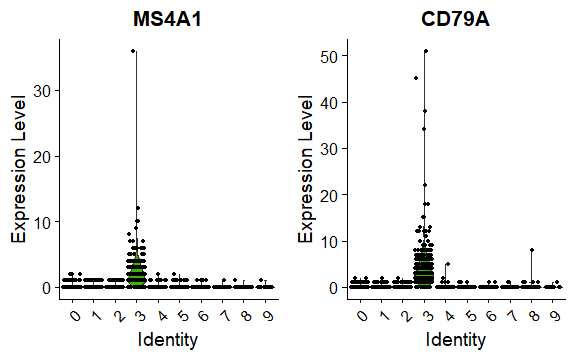
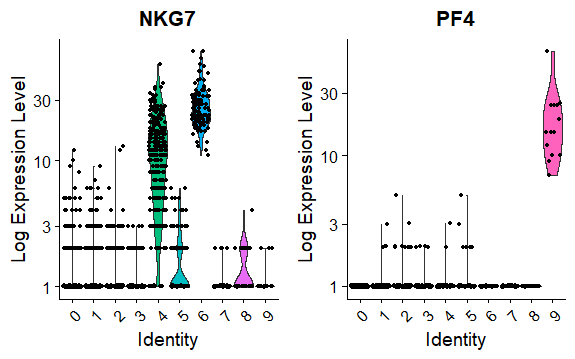
DNAproDB
DNA–protein complex structure analysis
Paper
Document
- Update
- Structure processing upgrades
- Data update
- Database and web interface
- Search
- Report
- Integration with Nucleic Acid Database
- Discussion and conclusion
- case study
EnhancerAtlas
Enhancers in nine species
Paper
Document
- Materals and methods
- Data sources
- Data processing for individual dataset/track
- Generation of consensus track
- Enhancer-gene interactions
- Implenentation of database
- Results
- statistics
- Database search
- Enhancer browser
GWAS Central
- Materials and methods
- Data collection and curation
- Ontology mapping
- Database design
- Results
- GWAS data searching
- Data visualization
- Discussion
MatrisomeDB
Extracellular matrix components
Paper
Document
- Database infrastructure
- Data sets included
- Mass spectrometry data search and processing
- Database features and functionalities
- MatrisomeDB query
- Hierarchically-clustered protein distribution heatmap
- Peptide converage map
- Results
- Experimental coverage of the in-silico predicted matrisome
- Building an ECM atlas
- Future directions
- Future data extension
- Extension to include studies from other model organisms
- Extracting quantitative data from label-free and label-based proteomics studies
MIBiG
Biosynthetic Gene Clusters of Known Function
Paper
Document: No
- Methods and implementation
- Manual curation of entries
- Data quality improvements
- The new database architecture
- Results and discussion
- Data overview
- BGC diversity
- Annotation completeness
- A new online repository
- Data overview
MirGeneDB
Animal miRNA complements
Paper
Document
- Expansion of MirGeneDB
- Quality of MirGeneDB annotations
- Improved web interface of MirGeneDB
- MiRNA nomenclature
- Functional classfication of MiRNA-seeds
- Future developments
MSDB
Microsatellites from all sequenced genomes
Paper
Document
- Materials and methods
- Data sources
- Identification of repeats
- Database design
- Web interface
- Database overview and functionality
- Dashbord view
- Species selection
- Modal view
- Tabel view
- Data download
- Help page
- Discussion
- Case study 1: length preference of AGAT repeats
- Case study 2: enrichment of AGC repeats in ruminants
Ohnologs
Vertebrate ohnologs
Paper
Document
- Results
- Data collection and processing
- Navigating teh OHNOLOGS database
- Summary of the contents of the OHNOLOGS database
PolyASite
RNA polyadenylation sites
Paper
Document
- Materials and methods
- Protocal-spefici pre-processing of reads
- Uniform analysis of putative poly(A) sites in individual samples
- Clustering of closely spaced sites
- Annotation and quantification of pply(A) sites
- Results
- Website roadmap
WALTZ-DB
Amyloidogenic peptide sequences
Paper
Document
- Expanded content and feature improvements
- New peptide entries and database statistics
- WALTZ-DB 2.0 novel features
- Materials and methods
- Peptide synthesis
- Determination of amyloid fibril properties
- Structural models and energy calculations
- Links to other databases