读入数据
基于上一篇Seurat的分群的结果进行处理,已经进行过预处理降维分群。
导入上篇保存的pbmc数据
1
> load('./output/res0.5.Robj')
取其中两个亚群进行分析
1
2
3> sub_pbmc <- subset(pbmc, idents=c(3,4))
# 目前数据是个seurat对象
> seurat_object <- sub_pbmc每个群取10个marker基因,为下面monocle分析准备
1
2> markers <- FindAllMarkers(sub_pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
> top10 <- markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_logFC)接下来从seurat object中提取创建monocle对象需要的数据
提取表达矩阵,转化为
matrix类型1
> data <- as(as.matrix(seurat_object@assays$RNA@data), 'sparseMatrix')
提取细胞表,并转化成
AnnotatedDataFrame类型1
> pd <- new('AnnotatedDataFrame', data = seurat_object@meta.data)
提取基因注释,并转化成
AnnotatedDataFrame类型1
2> fData <- data.frame(gene_short_name = row.names(data), row.names = row.names(data))
> fd <- new('AnnotatedDataFrame', data = fData)创建monocle对象
newCellDataSet需要输入一个表达矩阵,AnnotatedDataFrame格式的行坐标(sample name)和纵坐标(gene name)1
2
3
4
5
6#Construct monocle cds
> cds <- newCellDataSet(data,
phenoData = pd,
featureData = fd,
lowerDetectionLimit = 0.5,
expressionFamily = negbinomial.size());预估size factors和分散
1
2> cds <- estimateSizeFactors(cds)
> cds <- estimateDispersions(cds)提取表达基因
1
expressed_genes <- row.names(subset(fData(cds)))
建立轨迹
采用之前每个群选取的10个marker(top10)来建立轨迹
将top10读取成矩阵
1
2> ordering_genes<-as.matrix(top10)
ime.txt")过滤
1
> cds <- setOrderingFilter(cds, ordering_genes = ordering_genes)
降维
1
> cds <- reduceDimension(cds, max_components = 2, method = 'DDRTree',auto_param_selection = F) # take long time
排序
1
> cds <- orderCells(cds)
保存
1
2> save(cds,file="./output/orderCells.Robj")
write.table(pData(cds),file="./output/my_pseudotime.txt")查看
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15> head(pData(cds))
orig.ident nCount_RNA nFeature_RNA percent.mt RNA_snn_res.0.5 seurat_clusters S.Score G2M.Score
AAACATACAACCAC pbmc3k 2419 779 3.017776 4 4 0.06500339 -0.065012414
AAACATTGAGCTAC pbmc3k 4903 1352 3.793596 3 3 -0.01906540 -0.132349057
AAACCGTGTATGCG pbmc3k 980 521 1.224490 4 4 -0.01086219 0.005361576
AAACGCTGACCAGT pbmc3k 2175 782 3.816092 4 4 -0.03122878 -0.053972321
AAACGCTGGTTCTT pbmc3k 2260 790 3.097345 4 4 -0.03190767 -0.011423063
AAACTTGAAAAACG pbmc3k 3914 1112 2.631579 3 3 0.04395791 -0.080502879
Phase Size_Factor Pseudotime State
AAACATACAACCAC S 1.3046257 4.343051 6
AAACATTGAGCTAC G1 2.6443075 6.530045 8
AAACCGTGTATGCG G2M 0.5285379 0.000000 1
AAACGCTGACCAGT G1 1.1730306 3.882721 5
AAACGCTGGTTCTT G1 1.2188731 1.263559 1
AAACTTGAAAAACG S 2.1109157 6.543204 8画轨迹图
1
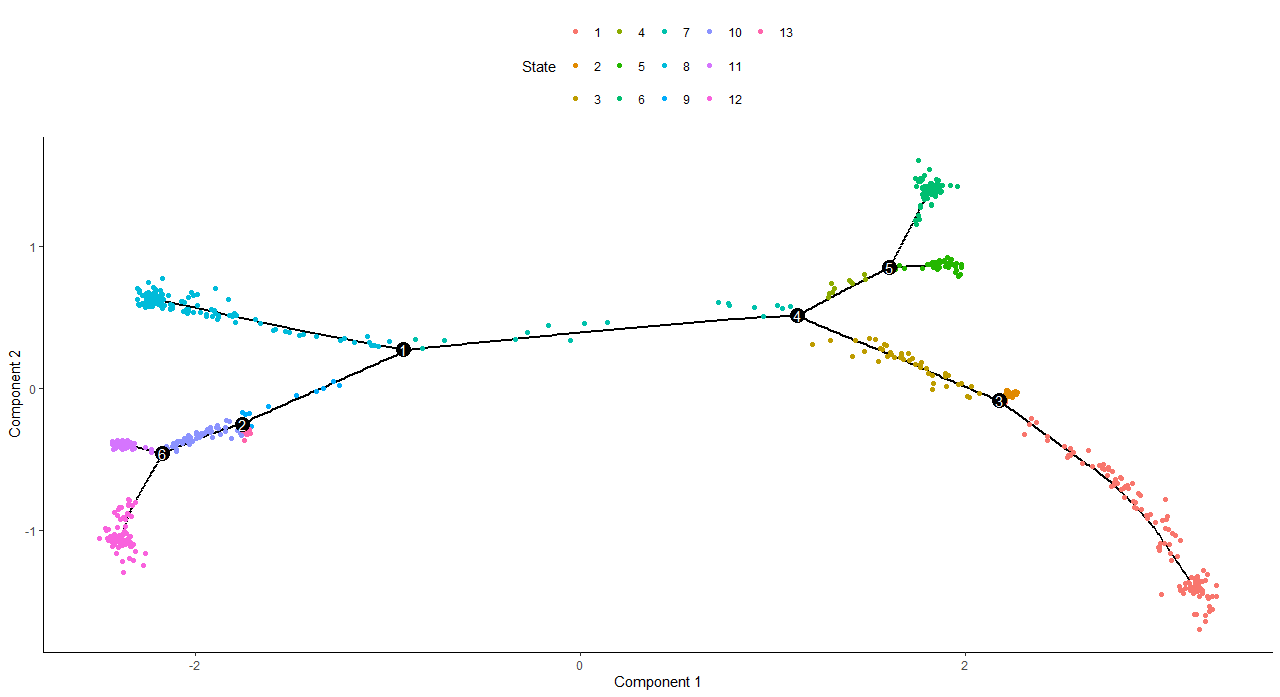
> plot_cell_trajectory(cds, color_by = "State")
1
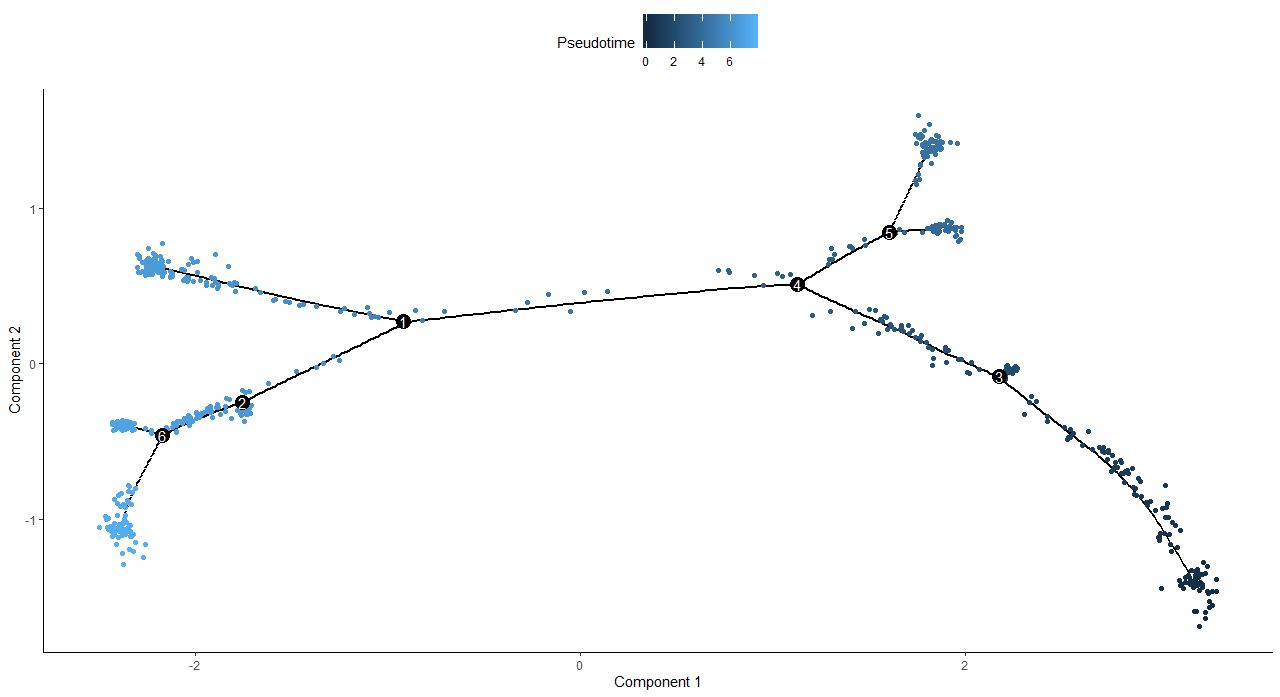
> plot_cell_trajectory(cds, color_by = "Pseudotime")
1
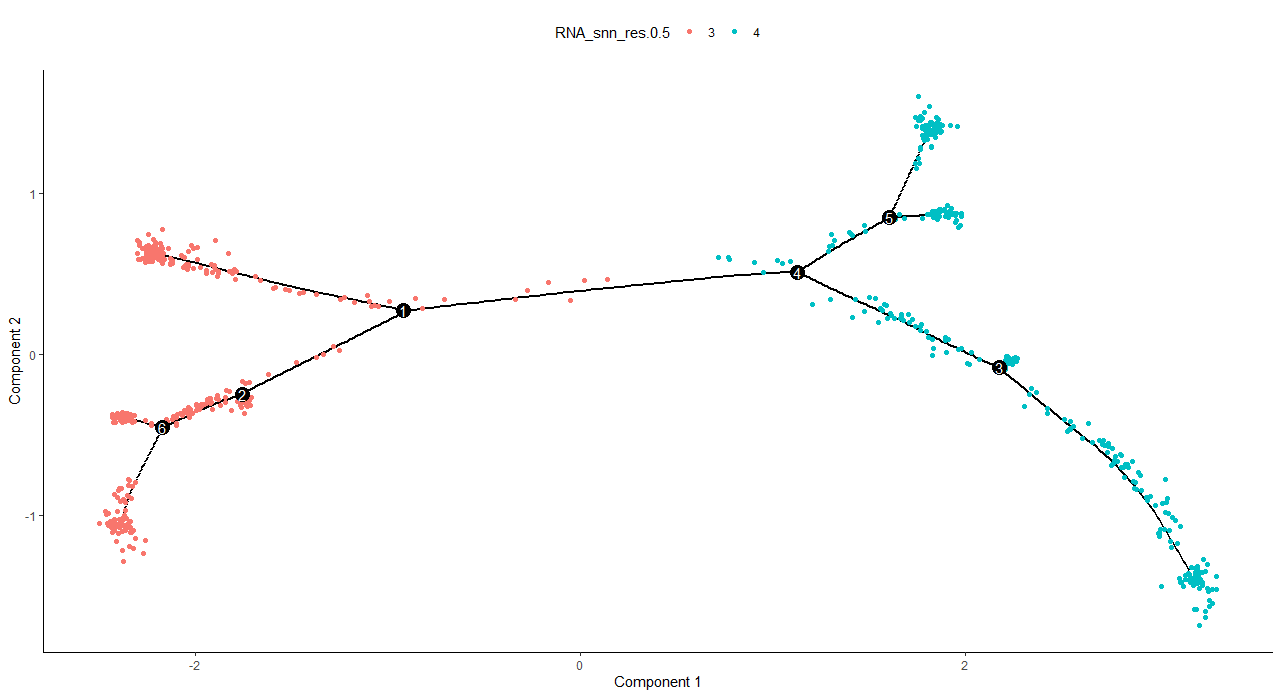
> plot_cell_trajectory(cds, color_by = "RNA_snn_res.0.5")
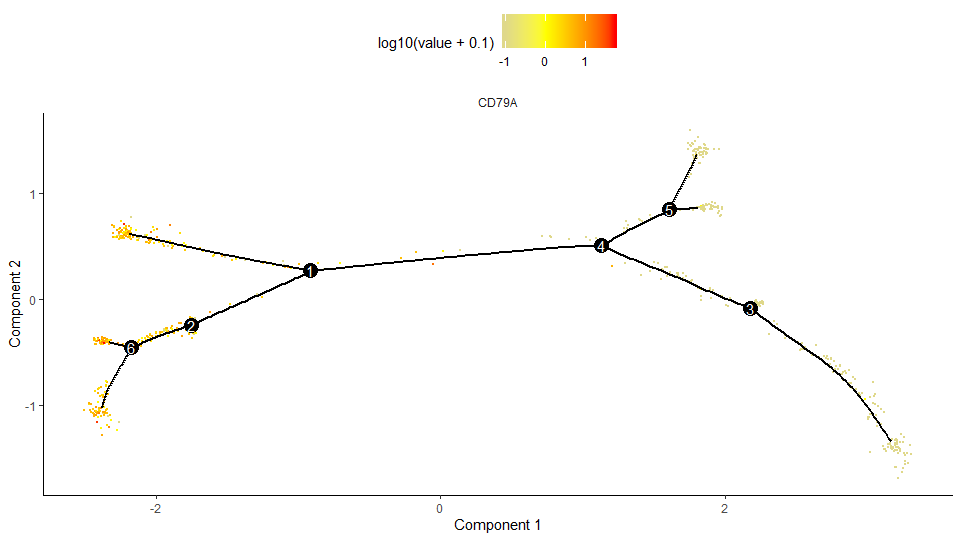
特定基因在在轨迹上的表达量图
1
> plot_cell_trajectory(cds, markers="CD79A", cell_size=0.5, use_color_gradient=T) + scale_color_gradient2(low="gray",mid="yellow",high="red")
展示感兴趣的基因热图(一个方向变化)
1
2
3
4
5> my_pseudotime_de <- differentialGeneTest(cds,fullModelFormulaStr = "~sm.ns(Pseudotime)",cores = 5)
> sig_gene_names <- row.names(subset(my_pseudotime_de, qval < 10^-20))
> plot_pseudotime_heatmap(cds[sig_gene_names,], num_clusters = 4,cores = 5,use_gene_short_name = TRUE,show_rownames = TRUE)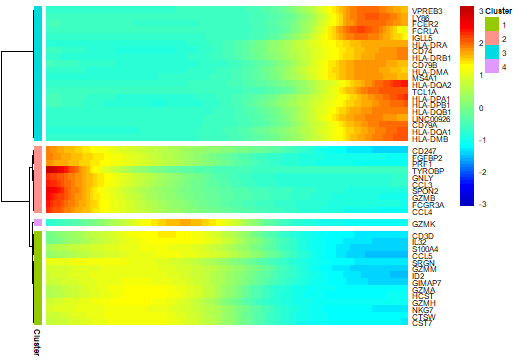
展示感兴趣基因基因拟时间表达变化(一个方向
1
2
3
4
5
6
7
8
9
10> c<-subset(my_pseudotime_de, qval < 10^-20)
> to_be_tested <- row.names(subset(fData(cds),gene_short_name %in% c("CD79A", "NKG7", "HLA-DRA", "CCL5")))
> cds_subset <- cds[to_be_tested,]
> diff_test_res <- differentialGeneTest(cds_subset,fullModelFormulaStr = "~sm.ns(Pseudotime)")
> diff_test_res[,c("gene_short_name", "pval", "qval")]
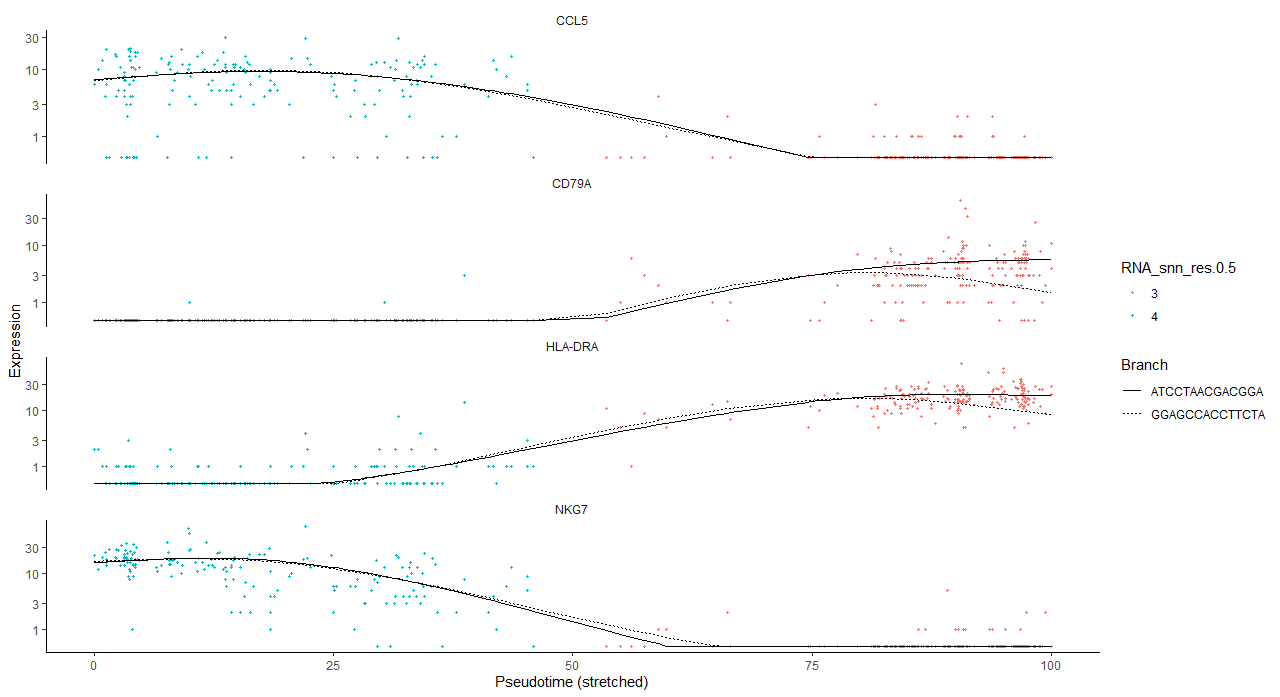
> plot_genes_in_pseudotime(cds_subset,color_by="RNA_snn_res.0.5")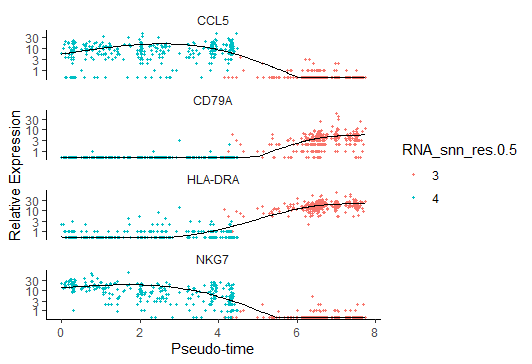
两个方向的图
热图
1
> plot_genes_branched_heatmap(cds[sig_gene_names,], num_clusters = 4,cores = 5,use_gene_short_name = TRUE,show_rownames = TRUE)
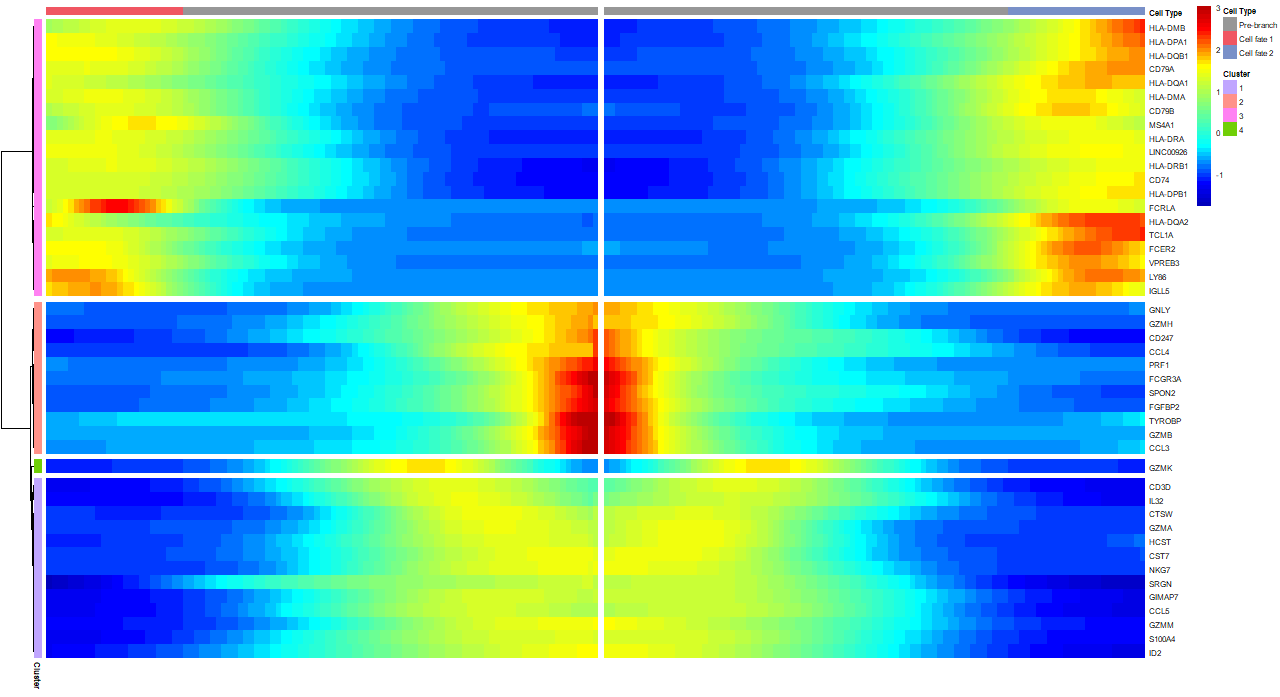
拟时间表达
1
> plot_genes_branched_pseudotime(cds_subset, branch_point = 2,color_by="RNA_snn_res.0.5")