前言
数据:10X官方提供的PBMC数据集,2700个外周血单核细胞(PBMC,Peripheral Blood Mononuclear Cells)公共数据,使用Illumina NextSeq 500测序。
原始教程和代码:Seurat - Guided Clustering Tutorial
1
2
3
4hg19
|__barcodes.tsv
|__genes.tsv
|__matrix.mtx数据读取
读入pbmc数据。
1
> pbmc.data <- Read10X(data.dir = "./data/pbmc3k_filtered_gene_bc_matrices/filtered_gene_bc_matrices/hg19/")
查看稀疏矩阵的维度,即基因数和细胞数。
1
2> dim(pbmc.data)
[1] 32738 2700查看矩阵的1-5行,1-3列。
“ · ”表示没有检测到,也就是0。这样表示是为了压缩空间。
1
2
3
4
5
6
7
8> pbmc.data[1:5,1:3]
5 x 3 sparse Matrix of class "dgCMatrix"
AAACATACAACCAC AAACATTGAGCTAC AAACATTGATCAGC
MIR1302-10 . . .
FAM138A . . .
OR4F5 . . .
RP11-34P13.7 . . .
RP11-34P13.8 . . .预处理:质控和数据筛选
创建Seurat对象
project是对象的名字,可以用来标识不同的样本
min.cell: 单个基因在小于等于3个细胞中测到就丢弃
min.features: 单个细胞测到小于等于200个基因说明可能是死细胞也丢弃
1
2
3
4> pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
An object of class Seurat
13714 features across 2700 samples within 1 assay
Active assay: RNA (13714 features)计算每个细胞的线粒体基因转录本数的百分比(%)
使用[[ ]] 操作符存放到metadata中
线粒体很多说明细胞可能是死细胞或者活性太低
1
> pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
检查一下metadata
查看metadata前三行
1
2
3
4
5> head(pbmc@meta.data, 3)
orig.ident nCount_RNA nFeature_RNA percent.mt
AAACATACAACCAC pbmc3k 2419 779 3.0177759
AAACATTGAGCTAC pbmc3k 4903 1352 3.7935958
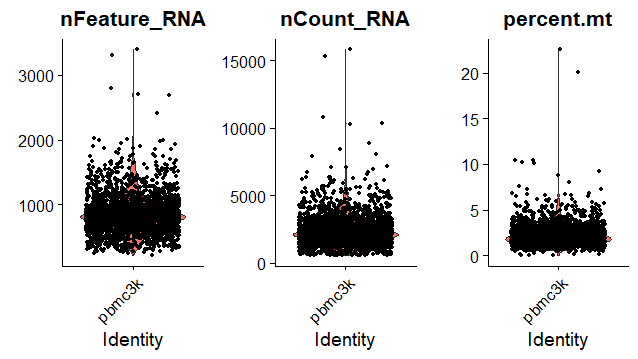
AAACATTGATCAGC pbmc3k 3147 1129 0.8897363展示基因及线粒体百分比
从图中得到过滤信息:观察数据的分布范围,异常数据要过滤掉
单个细胞基因数目nFeature_RNA:200-2500
线粒体占比percent.mt:小于5%
1
> VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
用散点图(FeatureScatter)来绘制两组feature信息的相关性,组合在一起(CombinePlots)显示
左图:reads count值和在线粒体的reads数量一般成反比,线粒体reads占比很高的情况一般有表达量的reads数很少
右图:基因(feature)数量一般会和reads count值成正比
1
2
3> plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
> plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
> CombinePlots(plots = list(plot1, plot2))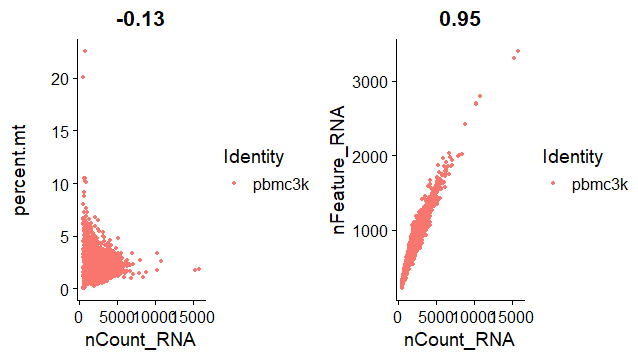
过滤细胞:
保留gene数大于200小于2500的细胞,目的是去掉空GEMs和1个GEMs包含2个以上细胞的数
保留线粒体基因的转录本数低于5%的细胞,目的是过滤掉死细胞等低质量的细胞数据。
1
> pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
表达量数据标准化
LogNormalize的算法:A = log( 1 + ( UMIA ÷ UMITotal ) × 10000 )
1
pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
差异表达分析
数据标准化:归一化
LogNormalize的算法:
A = log( 1 + ( UMIA ÷ UMITotal ) × 10000 )
得到的结果存储在:
pbmc[["RNA"]]@data1
2
3> pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
# 默认参数,所以省略也可以
# pbmc <- NormalizeData(pbmc)鉴定表达高变基因HVGs (highly variable features),
在细胞与细胞间进行比较,选择表达量差别最大的(也即是同一个基因在有的细胞中表达量很高,同时在部分细胞中表达很少)
利用
FindVariableFeatures函数,会计算一个mean-variance结果,也就是给出表达量均值和方差的关系并且得到top variable features2000个,用于下游分析,如PCA
1
> pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
提取表达量变变化最高的10个基因
1
2
3> top10 <- head(VariableFeatures(pbmc), 10)
> top10
[1] "PPBP" "LYZ" "S100A9" "IGLL5" "GNLY" "FTL" "PF4" "FTH1" "GNG11" "S100A8"画图
左图:把pbmc全部基因画出来,红色是2000个HVGs
右图:在左图基础上标出表达量变变化最高的10个基因
1
2
3plot1 <- VariableFeaturePlot(pbmc)
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)
CombinePlots(plots = list(plot1, plot2))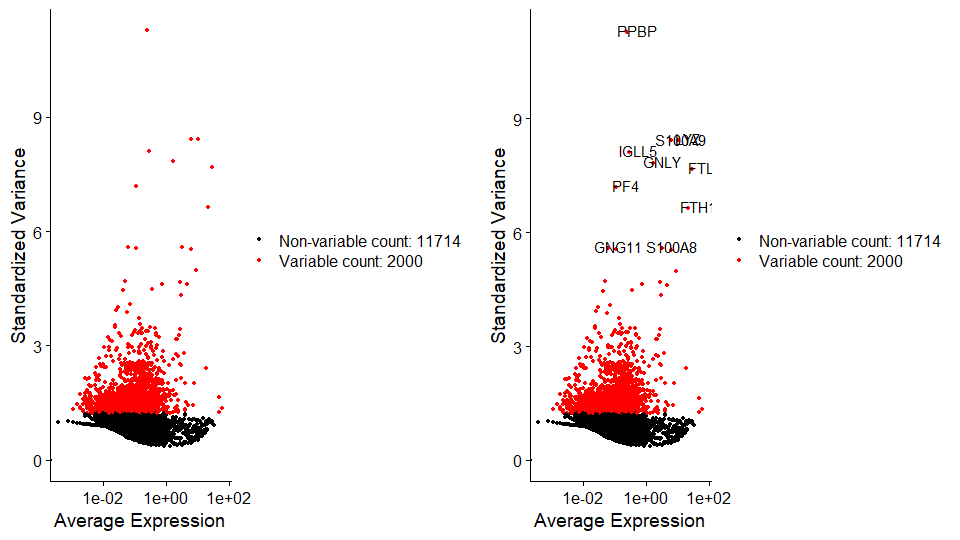
降维
数据标准化:零中心化
目的是对数据进行线性转换(scaling),是降维处理(如PCA)之前的标准预处理步骤。
使用ScaleData()函数:
center: 将每个基因在所有细胞的表达量都减去均值,使得每个基因在所有细胞的表达量均值为0
scale: 将center后每个基因的表达量除以标准差,让每个基因在所有细胞中的表达量方差为1
1
2
3
4
5## ScaleData()默认只是标准化高变基因(2000个),速度更快,不影响PCA和分群,但影响热图的绘制。
# pbmc <- ScaleData(pbmc,vars.to.regress ="percent.mt")
## 而对所有基因进行标准化的方法如下:
> all.genes <- rownames(pbmc)
> pbmc <- ScaleData(pbmc, features = all.genes)移除不想要的差异来源
如果要移除不想要的差异来源,可以用vars.to.regress
这个例子里移除了线粒体
更进一步的误差排除可以用另一个专业版函数:SCTransform,它也包含
vars.to.regress这个选项。1
> pbmc <- ScaleData(pbmc, vars.to.regress = "percent.mt")
线性降维:PCA
RunPCA:
默认input用高变基因集,但也可通过features参数自己指定
默认主成分数量是选50个
结果保存在reductions 这个接口中
1
> pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))
检查PCA分群结果
打印前5个PC,每个PC显示5个基因
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18## RunPCA结果保存在reductions这个接口中
# print(pbmc@reductions$pca, dims = 1:3, nfeatures = 5)
> print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)
PC_ 1
Positive: FTL, FTH1, CST3, AIF1, S100A4
Negative: MALAT1, IL32, LTB, IL7R, CCL5
PC_ 2
Positive: S100A8, S100A9, LYZ, CD14, LGALS2
Negative: B2M, NKG7, GZMA, CTSW, PRF1
PC_ 3
Positive: CD74, HLA-DRA, HLA-DPB1, HLA-DQA1, HLA-DQB1
Negative: PPBP, GNG11, SPARC, PF4, AP001189.4
PC_ 4
Positive: CD74, HLA-DQA1, HLA-DQB1, HLA-DQA2, HLA-DRA
Negative: FCGR3A, FTL, RP11-290F20.3, S100A4, NKG7
PC_ 5
Positive: FCGR3A, RP11-290F20.3, MS4A7, MALAT1, CKB
Negative: NKG7, LYZ, S100A9, S100A8, GNLY展示主成分基因分值
展示前两个PC里主成分基因的值
1
> VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")
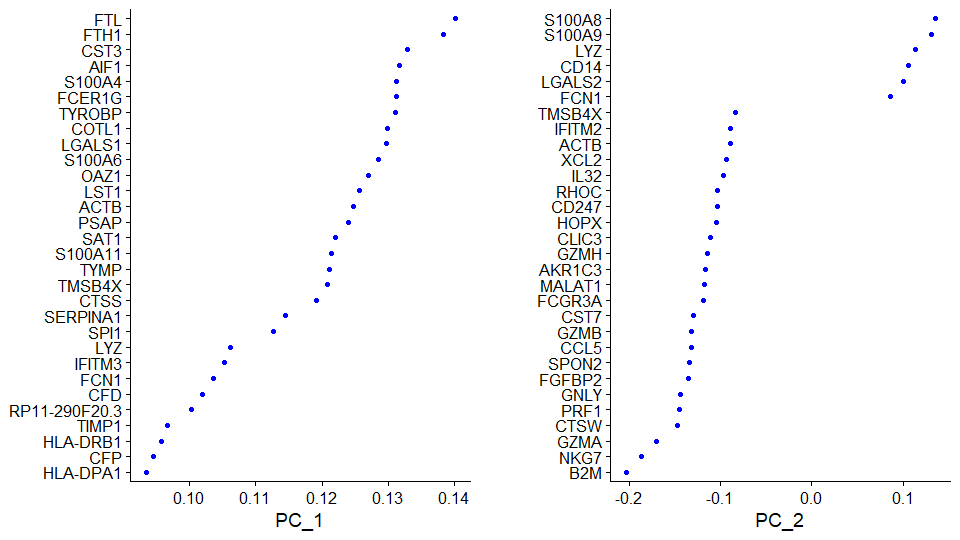
可视化降维后的结果
DimPlot:
默认提取前两个主成分,可以通过
dim选项修改,绘制散点图图中的每个点代表一个样本。
reduction指定降维方式类型,如果不指定,默认先搜索顺序umap->tsne->pca目前用的是PCA的结果
降维后的数据在
cell.embeddings中,可以打印出来1
2
3
4
5> DimPlot(pbmc, reduction = "pca")
> head(pbmc[['pca']]@cell.embeddings)[1:2,1:2]
PC_1 PC_2
AAACATACAACCAC -4.2153796 -0.8006517
AAACATTGAGCTAC 0.9503998 -3.0434884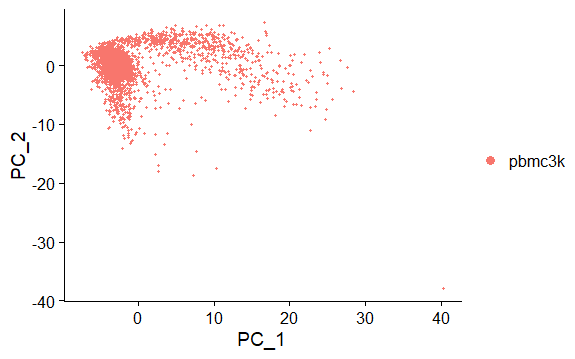
画第一个主成分的热图
默认画前30个基因,可以通过nFeatures设置
dim:选哪几个主成分
cells:细胞数
balanced:TRUE表示正负得分的基因各占一半
1
> DimHeatmap(pbmc, dims = 1, cells = 500, balanced = TRUE)
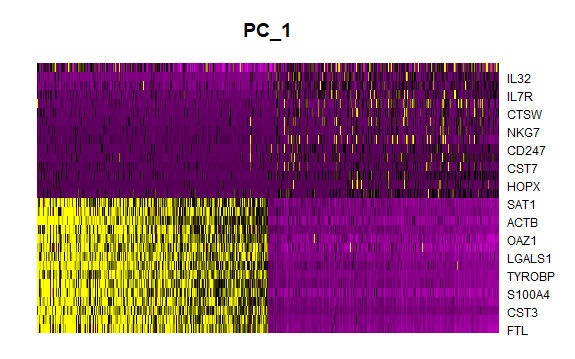
画第1到15个主成分的热图

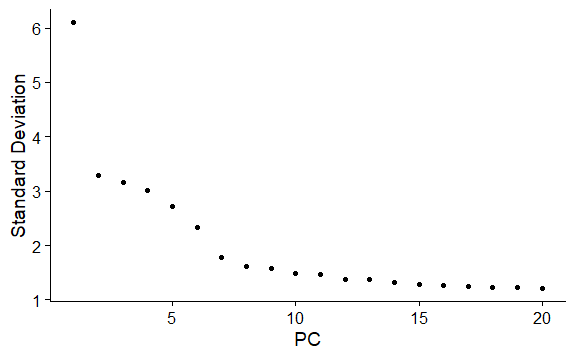
查看主成分贡献度
ElbowPlot画肘部图
主要关注”肘部“的PC,它是一个转折点,选用转折点之前的PC就可以比较好地代表总体变化。
从下图可以看出可以选择前10个PC
在选择此参数时,建议选择偏高的数字(为了获取更多的稀有分群,“宁滥勿缺”);有些亚群很罕见,如果没有先验知识,很难将这种大小的数据集与背景噪声区分开来。
非线性降维:UMAP/tSNE
性降维PCA(1933)一个特点就是:它将高维中不相似的数据点放在较低维度时,会尽可能多地保留原始数据的差异,因此导致这些不相似的数据相距甚远,大多的数据重叠放置
tSNE(2008)兼顾了高维降维+低维展示,降维后的那些不相似的数据点依然可以放得靠近一些,保证了点既在局部分散,又在全局聚集
UMAP(2018)相比tSNE又能展示更多的维度:参考文献Seurat里要用tSNE要先计算PCA。Seurat下游的分群只需要PCA降维结果就可以,但是用tSNE的话,二维显示的图比较好看。
Tsne降维并可视化
1
2
3
4
5
6> pbmc <- RunTSNE(pbmc, dims = 1:10)
> head(pbmc[['tsne']]@cell.embeddings)[1:2,1:2]
tSNE_1 tSNE_2
AAACATACAACCAC -10.99287 13.52608
AAACATTGAGCTAC -28.57216 -27.23769
DimPlot(pbmc,reduction = "tsne",label = TRUE,pt.size = 1.5)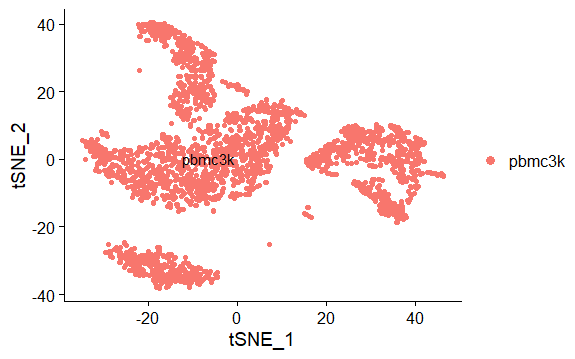Umap降维并可视化
1
2
3
4
5
6> pbmc <- RunUMAP(pbmc, dims = 1:10)
> head(pbmc[['umap']]@cell.embeddings)[1:2,1:2]
UMAP_1 UMAP_2
AAACATACAACCAC 3.784667 3.790803
AAACATTGAGCTAC 1.231294 -10.622349
> DimPlot(pbmc,reduction = "umap",label = TRUE,pt.size = 1.5)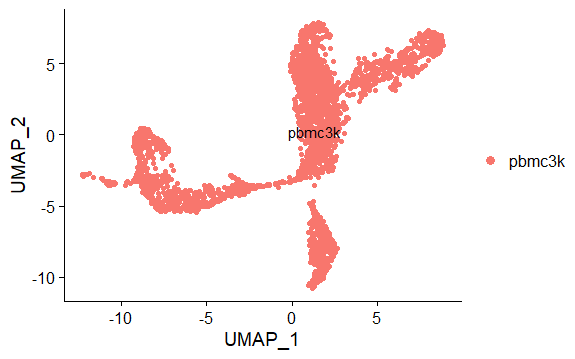
基于图的集群
计算权重
FindNeighbors是基于图的input是之前降维确定的主成分(这里选用了前10个)
根据PCA的结果,构建KNN图,得出近邻构成的小社区
在小社区内,计算细胞两两之间的联系数(shared overlap)得到每条边的权重(Jaccard similarity)
1
> pbmc <- FindNeighbors(pbmc, dims = 1:10)
聚类
FindClusters默认使用Louvain算法resolution参数决定下游聚类分析得到的分群数,对于3K左右的细胞,设为0.4-1.2 能得到较好的结果(官方说明);如果数据量增大,该参数也应该适当增大。
resolution可以理解为分辨率,如果分辨率越高,看得越清晰,分群数目就越多。
1
> pbmc <- FindClusters(pbmc, resolution = 0.5)
查看结果
Idents可以看出不同细胞的分群例子打印了前五个细胞结果
1
2
3AAACATACAACCAC AAACATTGAGCTAC AAACATTGATCAGC AAACCGTGCTTCCG AAACCGTGTATGCG
4 3 0 2 4
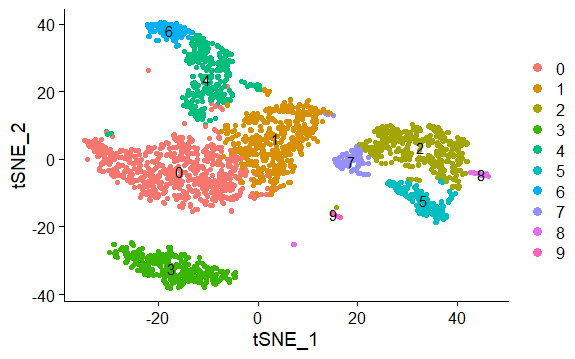
Levels: 0 1 2 3 4 5 6 7 8 9可视化
本例基于tsne降维结果,不同颜色表示不同的群
1
> DimPlot(pbmc,reduction = "tsne",label = TRUE,pt.size = 1.5)
识别差异表达基因(cluster biomarker)
识别cluster1中的marker基因
`FindAllMarkers()在细胞群之间比较,找到每个细胞群的正、负表达biomarker正负可以理解为上调、下调的意思;正marker表示在这个cluster中表达量高,而在其他的cluster中低;负marker表示在这个cluster中表达量低,而在其他的cluster中高
ident.1:第一个群ident.2:第二个群,如果不填就认为是除了第一个群之外的所有其他群的集合min.pct:基因在任何两群细胞中的占比最低不能低于多少其他可选参数,还有结果每列的含义等详见文档
1
2
3
4
5
6
7
8> cluster1.markers <- FindMarkers(pbmc, ident.1 = 1, min.pct = 0.25)
> head(cluster1.markers, n = 5)
p_val avg_logFC pct.1 pct.2 p_val_adj
CYBA 3.392038e-97 -50.92599 0.613 0.900 4.651841e-93
CD74 2.543403e-91 -203.51709 0.641 0.890 3.488023e-87
ACTB 1.818148e-84 -186.51617 0.982 0.995 2.493408e-80
OAZ1 2.698853e-79 -47.51709 0.777 0.943 3.701207e-75
FTH1 5.527059e-79 -370.51704 0.980 0.992 7.579808e-75识别每个cluster的maker基因,每个cluster取前两个maker保存到本地
1
2
3
4
5
6
7##寻找每一cluster的marker
> pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
##每组取前两个marker
> pbmc.markers.top <- pbmc.markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_logFC)
##存储marker
> write.table(pbmc.markers,file="./output/allmarker.txt")
> write.table(pbmc.markers.top,file="./output/topmarker.txt")比较cluster5和cluster0、cluster3的marker基因
ident.2 = c(0,3)说明把cluster0、cluster3合并作为一个群
1
2
3
4
5
6
7
8> cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, ident.2 = c(0, 3), min.pct = 0.25)
> head(cluster5.markers, n = 5)
p_val avg_logFC pct.1 pct.2 p_val_adj
CFD 1.444184e-208 12.930168 0.956 0.032 1.980554e-204
FCGR3A 1.905989e-199 20.996601 0.975 0.049 2.613873e-195
RP11-290F20.3 3.500106e-199 4.916265 0.875 0.018 4.800046e-195
IFITM3 1.752032e-198 49.819631 0.981 0.052 2.402737e-194
CD68 1.486609e-196 5.804065 0.950 0.042 2.038735e-192可视化
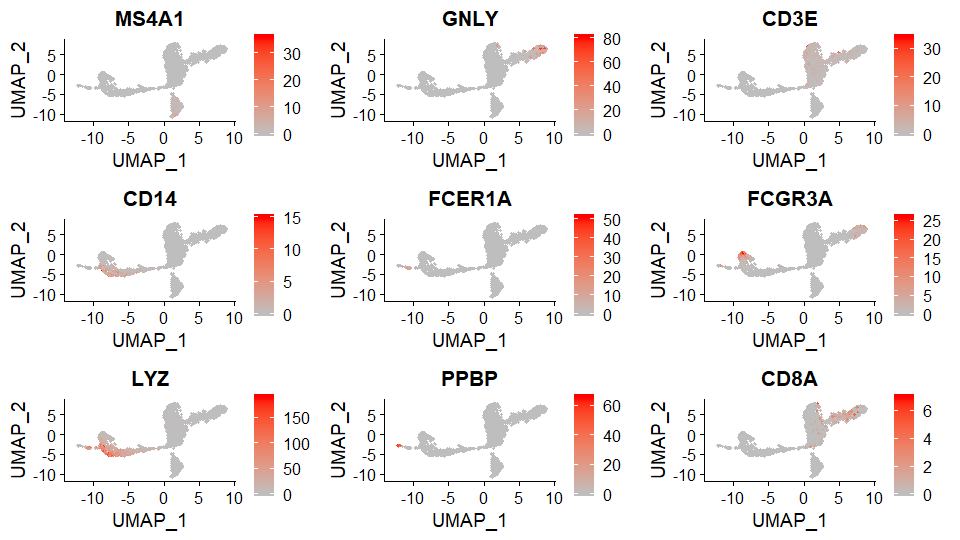
FeaturePlot最常用的可视化
将基因表达量投射到降维聚类结果中
1
> FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP", "CD8A"),cols = c("gray", "red"))
小提琴图
VlnPlot展示某些基因在所有细胞群的表达量
1
2> VlnPlot(pbmc, features = c("MS4A1", "CD79A"))
> VlnPlot(pbmc, features = c("NKG7", "PF4"), slot = "counts", log = TRUE)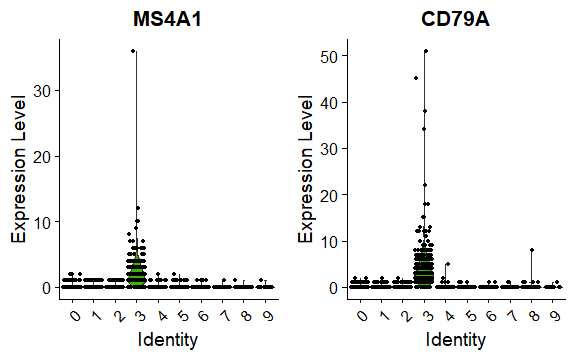
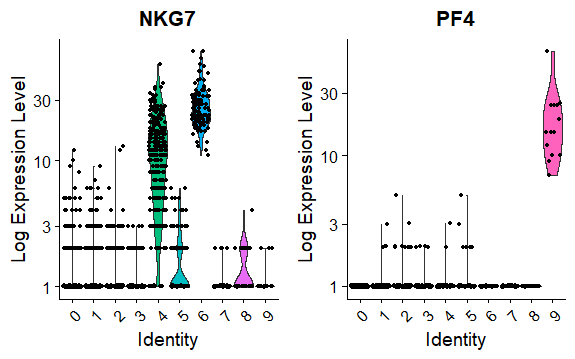
热图
DoHeatmap1
2
3## 每个细胞群选前十个基因,所以一共有100个基因
> top10 <- pbmc.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_logFC)
> DoHeatmap(pbmc, features = top10$gene) + NoLegend()
气泡图
DotPloty轴是细胞群
颜色表示平均表达量
大小表示表达的基因个数占总个数的百分比
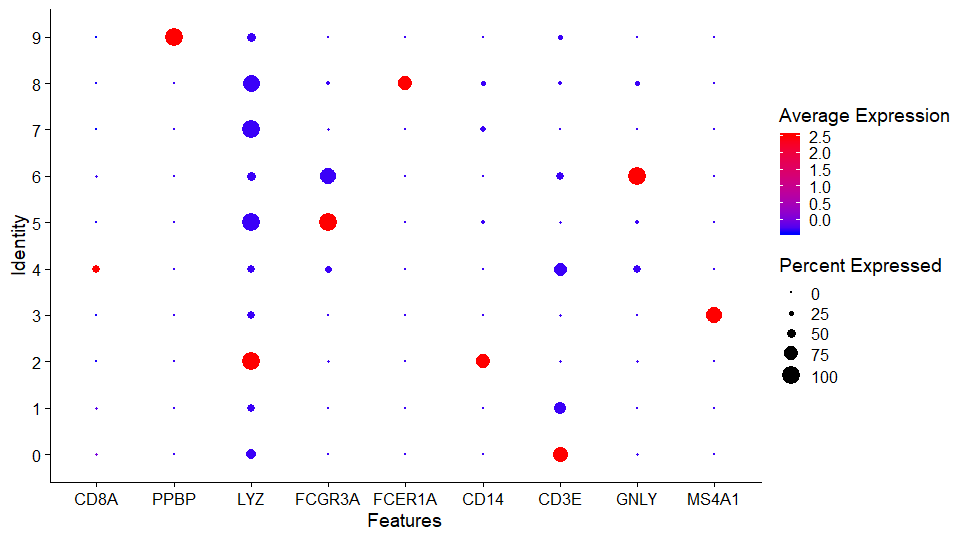
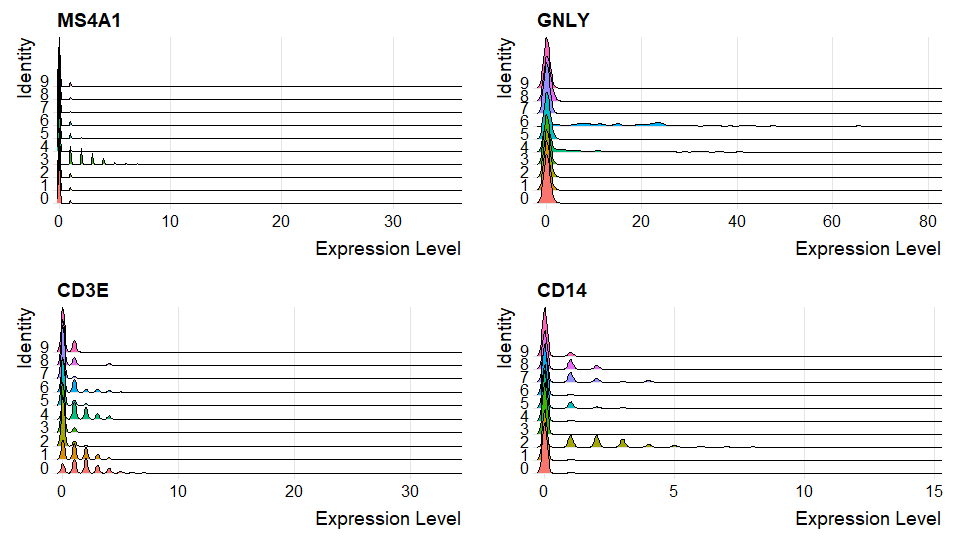
山脊图
RidgePlotx轴是表达量,y轴是细胞群
feature1的(x,y)位置山脊高说明feature1基因在y细胞群,表达量为x的细胞很多
1
RidgePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14"), ncol = 2)
判断细胞类型
根据每个群找出来的biomarker来判断每个细胞群的细胞类型。
需要结合额外的背景知识,可以查阅文献和数据库CellMarker。
官网给出的背景知识:
| Cluster ID | Markers | Cell Type |
| :————- | :—————— | :—————- |
| 0 | IL7R, CCR7 | Naive CD4+ T |
| 1 | IL7R, S100A4 | Memory CD4+ |
| 2 | CD14, LYZ | CD14+ Mono |
| 3 | MS4A1 | B |
| 4 | CD8A | CD8+ T |
| 5 | FCGR3A, MS4A7 | FCGR3A+ Mono |
| 6 | GNLY, NKG7 | NK |
| 7 | FCER1A, CST3 | DC |
| 8 | PPBP | Platelet |给群添加细胞类型标签(Cell Type)
1
2
3
4> new.cluster.ids <- c("Naive CD4 T", "Memory CD4 T", "CD14+ Mono", "B", "CD8 T", "FCGR3A+ Mono", "NK", "DC", "Platelet")
names(new.cluster.ids) <- levels(pbmc)
> pbmc <- RenameIdents(pbmc, new.cluster.ids)
> DimPlot(pbmc, reduction = "tsne", label = TRUE, pt.size = 1.5) + NoLegend()上面的代码是会报错的,因为我的分群结果是10个,但标签只有9个。
官网的分群结果是9个,背景知识表格给的也是9个,所以例子里的标签也是9个。
至于为什么一样的数据,一样的代码,一样的参数,结果确不一样呢?Who Knows.
不是真的研究数据所以不想做背景Research了,代码意会一下。
下一个教程会用一个自动注释的R包SingleR来添加标签。
保存数据
存储细胞归类结果
1
> saveRDS(pbmc, file = "./output/pbmc3k_final.rds")
Ref:
[1] 单细胞Seurat包升级之2,700 PBMCs分析(上)
[2] 单细胞Seurat包升级之2,700 PBMCs分析(下)
[3] Seurat - Guided Clustering Tutorial
[4] 一文读懂单细胞测序分析流程